Research Projects
Genomic underpinnings of exceptional traits in bats
The Hiller lab is a core member of the Bat1K initiative that aims at sequencing reference-quality genomes of all bats to unlock the genomic secrets behind their exceptional traits. Chiroptera (bats) are the only mammals that evolved powered flight and many bats possess echolocation to navigate and hunt. Bats exhibit an exceptional longevity among mammals, illustrated by the fact that 18 of the 19 mammals that have a longer body size-normalized lifespan than humans are bats. Bats are also reservoirs for many zoonotic viruses such as rabies, Ebola, and coronaviruses including SARS-CoV and SARS-CoV-2. However, while such viruses can be deadly for humans, viral infections in reservoir bats do not cause disease symptoms, indicating that they have a unique immune system [1].
In our previous work, we analyzed reference-quality genomes of six bat species that were sequenced by Bat1K, revealing loss and selection patterns in genes driving pro-inflammatory responses in the NF-κB pathway and expansion of antiviral defense pathways (APOBEC3 genes) [2].
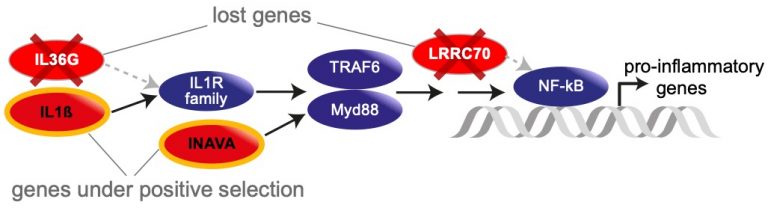
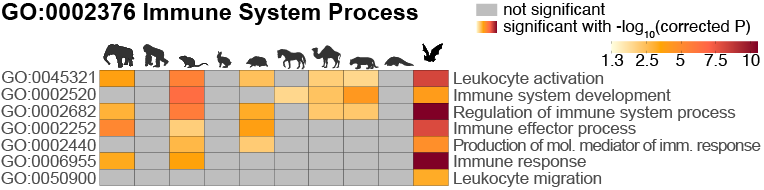
In our 2025 publication [3], we analyzed genomes of key coronavirus reservoir species (Rhinolophus and Hipposideros bats) to better understand the genomic basis of immunity-related changes. Our systematic analysis of 115 mammalian genomes revealed that bats harbor the highest prevalence of molecular signatures of immune adaptation among all mammals. Furthermore, the ancestral bat lineage shows a remarkable enrichment of positive selection in immune genes, providing genomic evidence that bats have pronounced immune system adaptations and indicating a connection to the evolution of flight.
Currently, as part of the ERC synergy project BatProtect, we are generating new high-quality genomes of ~150 bat species to uncover the genomic basis and evolution of extended healthspan and disease tolerance in bats.
[1] Wang et al. Decoding bat immunity: the need for a coordinated research approach. Nature Reviews Immunology, 21, 269–271, 2021
[2] Jebb et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature, 583, 578–584, 2020
[3] Morales et al. Bat genomes illuminate adaptations to viral tolerance and disease resistance. Nature, 637, 449–458, 2025
Genomics of dietary adaptations
Adaptations to specialized diets evolved many times independently in mammals and birds. For example, two groups of bats and several bird lineages (e.g. hummingbirds, sunbirds, honeyeaters) consume a sugar-rich diet consisting of fruit juice or nectar. Vampire bats are the only mammals that feed exclusively on blood. We are interested in uncovering the genomic underpinnings of metabolic adaptations to extreme diets that provide mainly sugar (fruit, nectar) or protein (blood) and ultimately aim at obtaining insights that could be ultimately harnessed for human metabolic disease therapy.
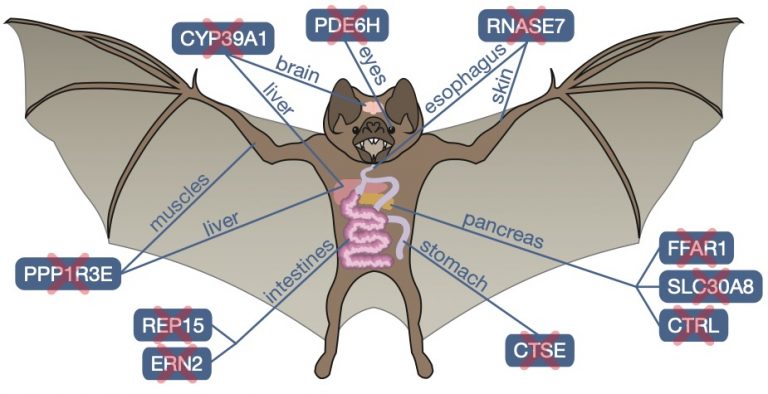
Our previous work discovered that two frugivorous bats lost genes that – when inactivated — enhance insulin secretion (FFAR3) and insulin sensitivity (FAM3B) [1]. Using our Forward Genomics framework we searched for genes preferentially lost in herbivorous and in carnivorous mammals [2]. For herbivores, our screen uncovered convergent losses of the lipase inhibitor PNLIPRP1, which enhances fat digestion, and repeated losses of the pancreatic exocytosis factor SYCN, which relates to continuous zymogen secretion. For carnivores, we found losses of genes regulating appetite and glucose homeostasis (INSL5, RXFP4) and losses of xenobiotic receptors (NR1I3, NR1I2) that reflecting irregular feeding patterns and a reduced xenobiotic content [2]. By generating a haplotype-resolved chromosome-level assembly of the common vampire bat, we discovered previously-unknown gene losses that relate to metabolic and physiological changes, including losses that likely contributed to enhanced iron excretion (CYP39A1) and the exceptional social behavior of vampire bats (REP15) [3]. We also discovered that hummingbirds lost a key enzyme, which facilitated metabolic muscle adaptations required for the evolution of hovering flight [4].
In a recent study [5], we generated new genomes and transcriptomes of birds that feed on sugar-rich nectar or fruit diets. Our integrative analysis revealed selection signals affecting genes and regulatory elements involved in sugar, lipid, and amino acid metabolism, as well as pathways associated in humans with type 2 diabetes and blood pressure regulation. We also detected strong evidence for convergent evolution, but lineage-specific changes also contribute to sugar diet adaptations. Highlighting a striking example of molecular convergence, the master regulator of sugar and lipid homeostasis MLXIPL (ChREBP) is the only gene with convergent adaptive evolution in all four sugar-feeding bird lineages, and experiments show that the hummingbird gene drives stronger upregulation of its target genes.
We are also studying vampire bats, the only tetrapods among >30,000 species that feed exclusively on blood. Through comparative analyses of new genomes and transcriptomes and experiments, we uncovered molecular underpinnings of their enlarged, non-acidic, absorptive stomach; altered gastrointestinal motility; trypsin-dependent protein digestion; upregulated amino acid catabolism; impaired digestion and increased de novo synthesis of fatty acids; defective sugar metabolism and natural insulin deficiency; enhanced heme iron absorption; and adult splenic erythropoiesis [6].
Currently, we are generating new genomes of Phyllostomid bats to investigate dietary diversification and the genetic adaptations underlying the extraordinary dietary diversity of this group [7].
[1] Sharma et al. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nature Communications, 9(1), 1215, 2018
[2] Hecker et al. Convergent gene losses illuminate metabolic and physiological changes in herbivores and carnivores. PNAS, 116:3036-3041, 2019
[3] Blumer et al. Gene losses in the common vampire bat illuminate molecular adaptations to blood feeding. Science Advances, 8 (12), eabm6494, 2022
[4] Osipova et al. Loss of a gluconeogenic muscle enzyme contributed to adaptive metabolic traits in hummingbirds. Science, 379(6628), 185-190, 2023
[5] Osipova et al. Convergent and lineage-specific genomic changes shape adaptations in sugar-consuming birds. Science, 391(6788), 2026
[6] Liu et al. Integrated genomics and transcriptomics reveal mechanisms of extreme dietary adaptation in vampire bats. submitted
[7] Yi et al. Comprehensive phylogenetic trait estimations support ancestral omnivory in the ecologically diverse bat family Phyllostomidae. Evolution, 79(11), 2406–2420, 2025
Adaptive gene losses
One would intuitively expect that inactivation (loss) of coding genes is often deleterious, after all loss-of-function mutations cause many human diseases. However, in evolution, gene loss can also contribute to adaptive traits (the “less is more” principle). We developed a computational method to accurately detect the gene losses across many species [1] (now incorporated in TOGA and TOGA2). Our research revealed a number of likely adaptive gene losses:
losses of epidermis-related genes (e.g. GSDMA, DSG4, DSC1, TGM5) in cetaceans likely contribute to the thick epidermis and loss of hair, representing adaptation to an aquatic environment [1],
loss of AMPD3 in sperm whale likely improves oxygen transport from lung to tissue, representing an advantage for a long-diving whale [1],
losses of renal transporter (e.g. SLC22A12 (URAT1), SLC2A9 (GLUT9), SLC22A6 (OAT1), RHBG), insulin secretion inhibitor (FFAR3) and pancreatic cytokine genes (FAM3B) likely help fruit bats to preserve precious electrolytes and enhance insulin secretion and insulin sensitivity [1],
convergent losses of MMP12, an elastin degrading protease, in cetaceans and manatees may be involved in “explosive exhalation”, a unique breathing adaptation [1],
repeated losses of the triglyceride lipase inhibitor PNLIPRP1 in herbivores is likely adaptive by enhancing the efficiency of triglyceride digestion [2],
a number of gene losses that occurred during the land to water transition in ancestral cetaceans are likely advantageous by reducing the risk of thrombus formation during diving (F12, KLKB1), improving the fidelity of DNA damage repair (POLM), and protecting from oxidative stress-induced lung inflammation (MAP3K19) [3],
in vampire bats, loss of REP15 could contribute to enhanced iron elimination (likely an adaptation to their iron-rich blood diet) and loss of CYP39A1 may have contributed to their exceptional social behavior, which is a unique feature among bats [4].
loss of the muscle-specific FBP2 gene in ancestral hummingbirds enhances
sugar metabolism and was likely a key step in the evolution hovering
flight [5].
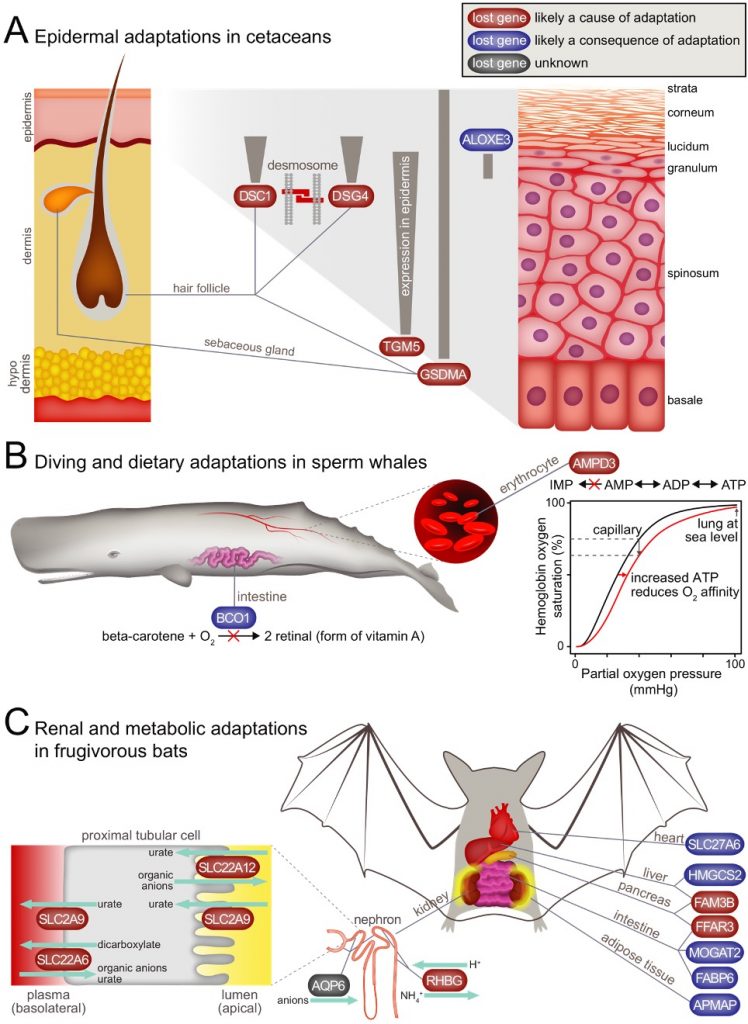
These results suggest that gene loss in mammals as an evolutionary mechanism for adaptation may be more widespread than previously thought. We continue to search for adaptive gene losses in mammals, birds and other organisms.
[1] Sharma et al. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nature Communications, 9(1), 1215, 2018
[2] Hecker et al. Convergent gene losses illuminate metabolic and physiological changes in herbivores and carnivores. PNAS, 116:3036-3041, 2019
[3] Huelsmann et al. Genes lost during the transition from land to water in cetaceans highlight genomic changes associated with aquatic adaptations. Science Advances, 5(9), eaaw6671, 2019
[4] Blumer et al. Gene losses in the common vampire bat illuminate molecular adaptations to blood feeding. Science Advances, 8 (12), eabm6494, 2022
[5] Osipova et al. Loss of a gluconeogenic muscle enzyme contributed to adaptive metabolic traits in hummingbirds. Science, 379(6628), 185-190, 2023
Forward Genomics: Linking phenotypic to genomic changes across species
Understanding which genomic changes underlie particular phenotypic changes between species is challenging, but a central goal in genomics and evolutionary biology. We developed the „Forward Genomics“ framework that focuses on independently evolved phenotypic differences and uses ancestral sequence reconstruction to systematically search whole genomes for differences that match the given phenotypic pattern [1].
We have improved the original prototype method by taking species relatedness and differences in evolutionary rates into account and computing P-values for phenotype-genotype associations [2]. To improve the power to detect relevant changes in cis-regulatory elements, we developed “Regulatory Element forward genomics” (REforge) and “Transcription factor forward genomics” (TFforge) that measure transcription factor binding site divergence across a phylogeny and detect both regulatory elements and regulators (TFs) that are associated with a given phenotype [3,4]. We also developed Forward Genomics methods that specifically associate gene losses with phenotypes [5].
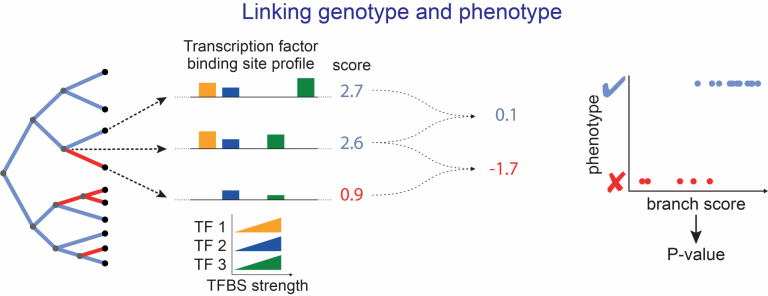
Applying these methods to traits such as vitamin C synthesis [1], eye degeneration in subterranean and other mammal with poor vision [6,10] loss of limb in snakes and limbless reptiles [6,7], an aquatic lifestyle [5], loss of teeth [5], loss of the vomeronasal system [8] and dietary adaptations [9] revealed insights into which genes and regulatory elements are associated with these phenotypic changes. Our recent review summarizes the latest developments and highlights key insights gained with Forward Genomics-like approaches [11].
[1] Hiller et al. A “forward genomics” approach links genotype to phenotype using independent phenotypic losses among related species. Cell Reports, 2(4), 817-823, 2012
[2] Prudent et al. Controlling for phylogenetic relatedness and evolutionary rates improves the discovery of associations between species’ phenotypic and genomic differences. Mol Biol Evol, 33(8), 2135-50, 2016
[3] Langer et al. REforge associates transcription factor binding site divergence in regulatory elements with phenotypic differences between species. Mol Biol Evol, 35(12), 3027–3040, 2018
[4] Langer & Hiller. TFforge utilizes large-scale binding site divergence to identify transcriptional regulators involved in phenotypic differences. Nucleic Acids Res, 47(4) e19, 2018
[5] Sharma et al. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nature Communications, 9(1), 1215, 2018
[6] Roscito et al. Phenotype loss is associated with widespread divergence of the gene regulatory landscape in evolution. Nature Communications, 9:4737, 2018
[7] Roscito et al. Convergent and lineage-specific genomic differences in limb regulatory elements in limbless reptile lineages. Cell Reports, 38(3):110280, 2022
[8] Hecker et al. Convergent vomeronasal system reduction in mammals coincides with convergent losses of calcium signaling and odorant degrading genes. Molecular Ecology, 28(16), 3656-3668, 2019
[9] Hecker et al. Convergent gene losses illuminate metabolic and physiological changes in herbivores and carnivores. PNAS, 116(8), 3036-3041, 2019
[10] Indrischek et al. Vision-related convergent gene losses reveal SERPINE3’s unknown role in the eye. Elife, 11:e77999, 2022
[11] Hilgers & Hiller. Linking phenotype to genotype using comprehensive genomic comparisons. Current Opinion in Genetics & Development, 94:102384, 2025
Development of comparative genomics methods
The Hiller lab has a long-standing interest in developing new genomics methods. Over the last years, we have established a powerful toolbox to address the phenotype – genotype question. This includes methods to detect functionally-relevant genomic changes in genes [1,2] and cis-regulatory elements [3,4] and improved methods to discover statistical associations between genomic and phenotypic changes [5].
Since high-quality whole genome alignments are the basis for our comparative analyses, we not only generate large alignments of vertebrates and mammals [6,7] but also developed methods that improve alignment sensitivity by detecting novel alignments in repeat-masked regions [8] and methods that improve alignment specificity by removing random alignments [9].
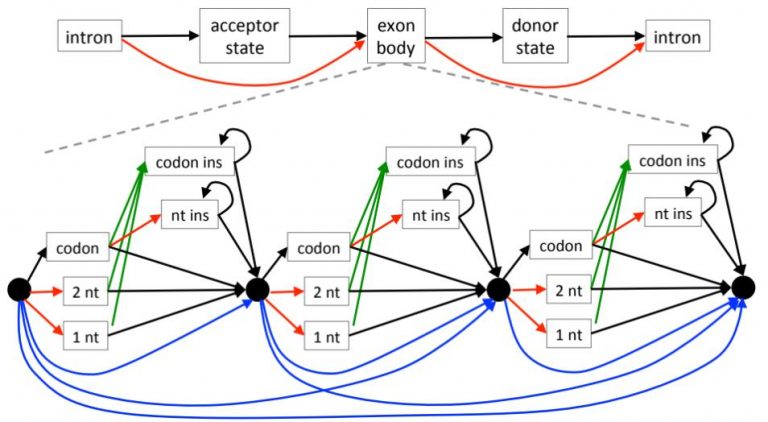
Annotating genes and inferring orthologs are a classical challenges in genomics. We developed TOGA (Tool to infer Orthologs from Genome Alignments), the first method that integrates gene annotation and ortholog identification [10]. TOGA annotates genes with our Hidden Markov Model based method CESAR [11,12] and implements a novel paradigm to detect orthologous gene loci that relies on machine learning and capturing intronic/intergenic alignments. TOGA is a scalable approach, allowing us to annotate and infer orthologs for >500 mammal and bird genomes.
Currently, we are finalizing TOGA2, the next-generation version of the TOGA framework, and provide annotations, orthologs, gene/loss duplication events and more for thousands of vertebrate genomes [13]. We are also developing more accurate methods to generate multiple codon alignments, to automate selection screens, and to detect additional types of functionally-relevant genomic changes.
[1] Sharma et al. A genomics approach reveals insights into the importance of gene losses for mammalian adaptations. Nature Communications, 9(1), 1215, 2018
[2] Lee et al. Molecular parallelism in fast-twitch muscle proteins in echolocating mammals. Science Advances, 4(9), eaat9660, 2018
[3] Langer et al. REforge associates transcription factor binding site divergence in regulatory elements with phenotypic differences between species. Mol Biol Evol, 35(12), 3027–3040, 2018
[4] Roscito et al. Convergent and lineage-specific genomic differences in limb regulatory elements in limbless reptile lineages. Cell Reports, 38(3):110280, 2022
[5] Prudent et al. Controlling for phylogenetic relatedness and evolutionary rates improves the discovery of associations between species’ phenotypic and genomic differences. Mol Biol Evol, 33(8), 2135-50, 2016
[6] Hecker & Hiller. A genome alignment of 120 mammals highlights ultraconserved element variability and placenta associated enhancers. GigaScience, 9(1), giz159, 2020
[7] Sharma & Hiller. Increased alignment sensitivity improves the usage of genome alignments for comparative gene annotation. Nucleic Acids Res, 45(14), 8369–8377, 2017
[8] Osipova et al. RepeatFiller newly identifies megabases of aligning repetitive sequences and improves annotations of conserved non-exonic elements. GigaScience, 8(11), giz132, 2019
[9] Suarez et al. chainCleaner improves genome alignment specificity and sensitivity. Bioinformatics, 33(11), 1596-1603, 2017
[10] Kirilenko et al. Integrating gene annotation with orthology inference at scale. Science, 380 (6643), 2023
[11] Sharma et al. Coding Exon-Structure Aware Realigner (CESAR) utilizes genome alignments for accurate comparative gene annotation. Nucleic Acids Res, 44(11), e103, 2016
[12] Sharma et al. CESAR 2.0 substantially improves speed and accuracy of comparative gene annotation. Bioinformatics, 33(24), 3985–3987, 2017
[13] Malovichko et al. Accurate, comprehensive gene annotation and ortholog identification across thousands of vertebrate genomes with TOGA2. in prep